Information about the Rudolph Lab.
 The Rudolph Lab is developing and studying genetic mouse models relevant for pathophysiology and treatment of psychiatric disorders using molecular biological, biochemical, morphological, optogenetic, chemogenetic and behavioral methods. A major focus is on synaptic and extrasynaptic inhibition in the CNS, specifically the physiological and pharmacological functions of inhibitory neurotransmission viaGABAA receptor subtypes and of excitatory neurotransmission via NMDA receptors.
The Rudolph Lab is developing and studying genetic mouse models relevant for pathophysiology and treatment of psychiatric disorders using molecular biological, biochemical, morphological, optogenetic, chemogenetic and behavioral methods. A major focus is on synaptic and extrasynaptic inhibition in the CNS, specifically the physiological and pharmacological functions of inhibitory neurotransmission viaGABAA receptor subtypes and of excitatory neurotransmission via NMDA receptors.
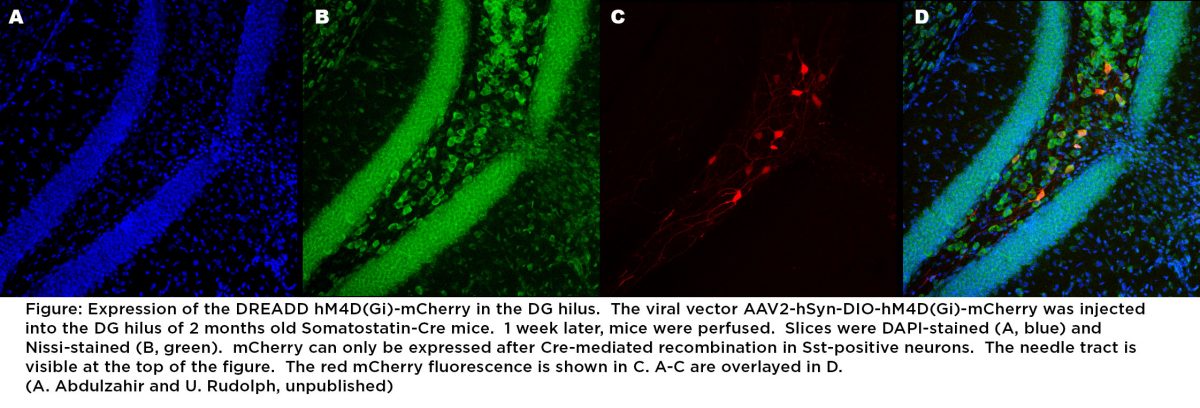
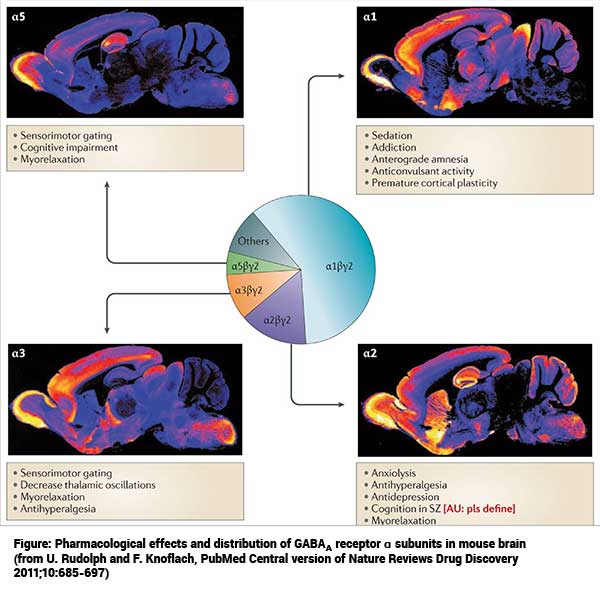
GABAA receptors are molecular substrates for the regulation of vigilance, anxiety, fear, sensorimotor gating, and learning and memory. Using knock-in point mutations in mice, we were able to demonstrate that anxiolytic and sedative actions of benzodiazepines like diazepam are mediated by distinct GABAA receptor subtypes and can thus be pharmacologically separated, which is of relevance for drug development (U. Rudolph et al., Nature 1999;401:796-800; K. Low et al., Science 2000;290:131-134, U. Rudolph and F. Knoflach, Nature Reviews Drug Discovery 2011;10:685-697). Using conditional gene targeting, we found that anxiety and fear are modulated by distinct intrahippocampal circuits, and that tonic inhibitory control of the dentate gyrus granule cells reduces memory interference (E. Engin et al., Journal of Neuroscience 2015;35:13698-13712; E. Engin et al., eLife 2016;5:e14120; Engin et al., Trends in Pharmacological Sciences 2018;39:710-732). We also interested in the role of specific GABAA receptors in the response to chronic social defeat stress and thus potentially for the development of depression.
Our Principal Investigator is Uwe Rudolph.
Click the tabs below or visit our website here to learn more.
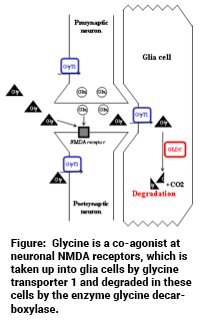 Structural variants like copy number variations (CNV), i.e., genomic deletions and duplications, have been discovered as rare mutations with sometimes large effect sizes in psychotic disorders. In a family affected by schizoaffective disorder and bipolar disorder with psychotic features, a small supernumerary marker chromosome containing genomic material from the 9p24.1 region was identified, that segregates with disease (J. TCW et al., Stem Cell Reports 2017;8:519-528; C.M. Grochowski et al., Human Mutation 2018;39:939-946). This results in a “duplication” (i.e., 3 copies instead of 2 copies) of some genes in this genomic region and a “triplication” (i.e., 4 copies instead of 2 copies) of the GLDC gene encoding glycine decarboxylase. Glycine decarboxylase degrades glycine, which is a co-agonist at the NMDA receptor. NMDA receptor hypofunction has been postulated to be a major pathophysiological factor in schizophrenia. Our working hypothesis is that the increased GLDC copy number results in increased amounts of the enzyme glycine decarboxylase, which then results in reduced glycine levels in astrocytes, from which it is known to be released by afferent innervation via AMPA receptors. This would then result in decreased availability of glycine at neuronal NMDA receptors and thus NMDA receptor hypofunction.
Structural variants like copy number variations (CNV), i.e., genomic deletions and duplications, have been discovered as rare mutations with sometimes large effect sizes in psychotic disorders. In a family affected by schizoaffective disorder and bipolar disorder with psychotic features, a small supernumerary marker chromosome containing genomic material from the 9p24.1 region was identified, that segregates with disease (J. TCW et al., Stem Cell Reports 2017;8:519-528; C.M. Grochowski et al., Human Mutation 2018;39:939-946). This results in a “duplication” (i.e., 3 copies instead of 2 copies) of some genes in this genomic region and a “triplication” (i.e., 4 copies instead of 2 copies) of the GLDC gene encoding glycine decarboxylase. Glycine decarboxylase degrades glycine, which is a co-agonist at the NMDA receptor. NMDA receptor hypofunction has been postulated to be a major pathophysiological factor in schizophrenia. Our working hypothesis is that the increased GLDC copy number results in increased amounts of the enzyme glycine decarboxylase, which then results in reduced glycine levels in astrocytes, from which it is known to be released by afferent innervation via AMPA receptors. This would then result in decreased availability of glycine at neuronal NMDA receptors and thus NMDA receptor hypofunction.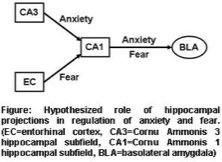 We have previously shown that while pharmacological reduction of anxiety (by the benzodiazepines diazepam) is dependent on α2-containing GABAA receptors in dentate gyrus granule cells and CA3 pyramidal neurons, pharmacological reduction of fear is dependent on α2-containing GABAA receptors in CA1 pyramidal neurons (E. Engin et al., eLife 2016;5:e14120; Engin et al., Trends in Pharmacological Sciences 2018;39:710-732). Our working hypothesis is that distinct intrahippocampal projections mediate anxiety and fear, with unique roles of the monosynaptic and the trisynaptic hippocampal pathways. We now want to determine how individual projections contribute to the bidirectional modulation of anxiety and fear using optogenetic approaches.
We have previously shown that while pharmacological reduction of anxiety (by the benzodiazepines diazepam) is dependent on α2-containing GABAA receptors in dentate gyrus granule cells and CA3 pyramidal neurons, pharmacological reduction of fear is dependent on α2-containing GABAA receptors in CA1 pyramidal neurons (E. Engin et al., eLife 2016;5:e14120; Engin et al., Trends in Pharmacological Sciences 2018;39:710-732). Our working hypothesis is that distinct intrahippocampal projections mediate anxiety and fear, with unique roles of the monosynaptic and the trisynaptic hippocampal pathways. We now want to determine how individual projections contribute to the bidirectional modulation of anxiety and fear using optogenetic approaches.