Information about the Chung Lab.
 The Chung lab seeks to have a better understanding of the pathogenesis of epilepsy. Understanding the pathogenesis is critical to developing novel therapeutic interventions and early diagnostics for epilepsy. “How does a neuron change itself to produce excessive electrical signals (intrinsic excitability) and how do neurons change their strength in communication (synaptic transmission) in an epileptic brain compared to a normal brain?”
The Chung lab seeks to have a better understanding of the pathogenesis of epilepsy. Understanding the pathogenesis is critical to developing novel therapeutic interventions and early diagnostics for epilepsy. “How does a neuron change itself to produce excessive electrical signals (intrinsic excitability) and how do neurons change their strength in communication (synaptic transmission) in an epileptic brain compared to a normal brain?”
The clue comes from the fact that inherited and de novo epilepsy is associated with mutations in ion channels, which are pore-forming proteins that generate electric current by mediating the flow of ions across the plasma membrane. They regulate electrical signals in morphologically and functionally distinct neuronal compartments: axons and dendrites. Axons deliver electrical signals to other neurons by action potentials whereas dendrites receive them at intercellular junctions called synapses. Ion channels enriched in axons are important for action potential initiation and termination, whereas ion channels enriched at synapses such as glutamate receptors are important for communication between neurons.
Since ion channels are critical regulators of neuronal activity, the two major long-term goals of Chung lab research have been to:
- Understand how epilepsy mutations affect ion channel function and lead to neuronal hyperactivity in inherited or de novo epilepsy.
- Identify molecular mechanisms that persistently alter ion channel function to cause hyperactivity during the development of acquired epilepsy.
To achieve these goals, Chung lab employs interdisciplinary approaches including primary neuronal culture, microscopy, biochemistry, electrophysiology and transgenic mice.
Our Principal Investigator is Hee Jung Chung.
Click the tabs below to learn more about our research and check us out at our website here!
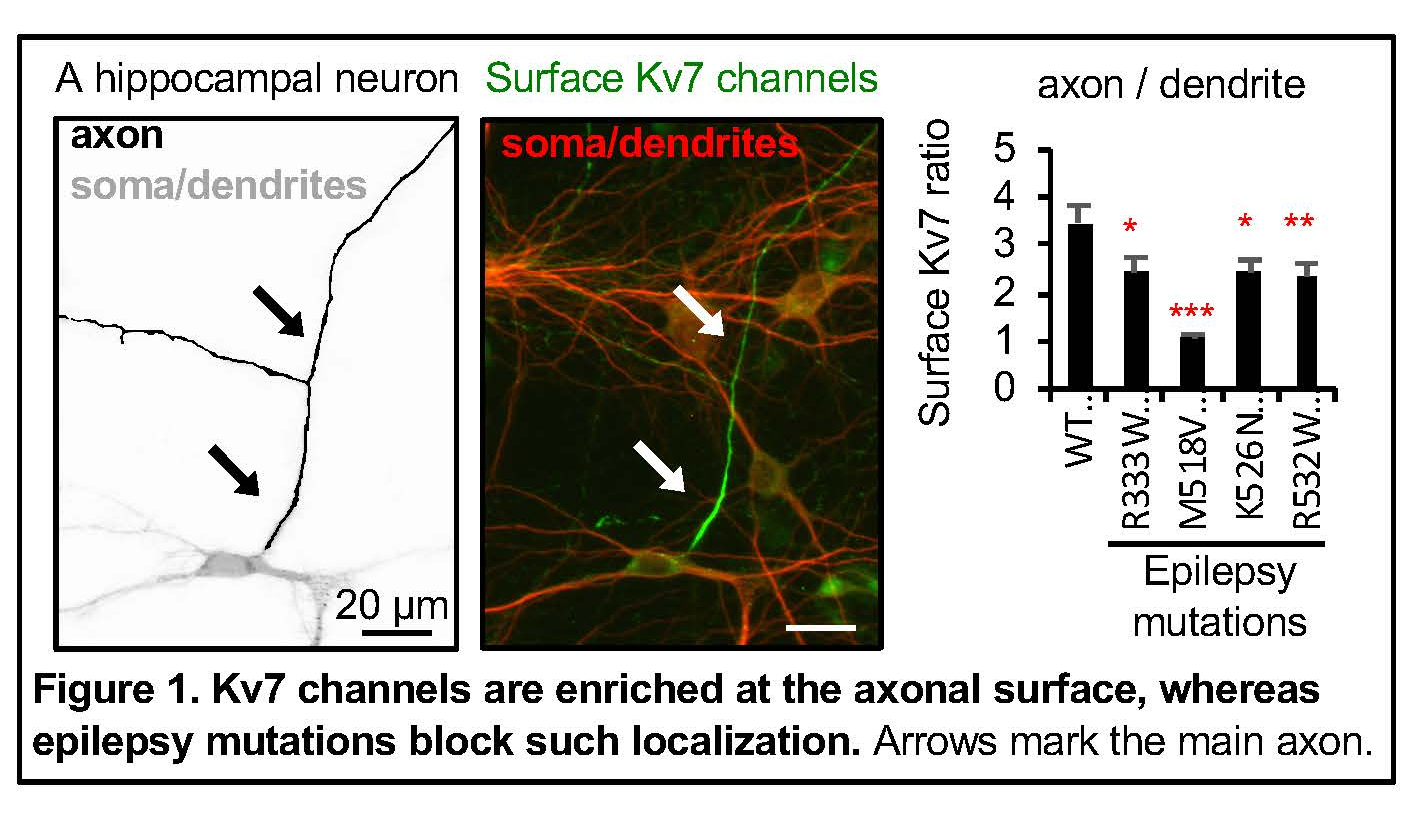 Neuronal Kv7/KCNQ potassium channels are heterotetramers composed of Kv7.2/KCNQ2 and Kv7.3/KCNQ3 subunits (Brown and Passmore, 2009), which are found throughout the brain including the hippocampus and neocortex (Cooper et al., 2001; Pan et al., 2006). Highly concentrated at the axonal initial segments (Pan et al., 2006) and preferentially enriched at the axonal plasma membrane compared to somatodendritic surface (Chung et al., 2006), these channels give rise to slowly activating and non-inactivating voltage-dependent outward potassium currents which potently inhibit both the repetitive and the burst firing of action potentials (Brown and Passmore, 2009).
Neuronal Kv7/KCNQ potassium channels are heterotetramers composed of Kv7.2/KCNQ2 and Kv7.3/KCNQ3 subunits (Brown and Passmore, 2009), which are found throughout the brain including the hippocampus and neocortex (Cooper et al., 2001; Pan et al., 2006). Highly concentrated at the axonal initial segments (Pan et al., 2006) and preferentially enriched at the axonal plasma membrane compared to somatodendritic surface (Chung et al., 2006), these channels give rise to slowly activating and non-inactivating voltage-dependent outward potassium currents which potently inhibit both the repetitive and the burst firing of action potentials (Brown and Passmore, 2009).